Bệnh Huntington: Căn bệnh khiến con người múa giật không kiểm soát
Bệnh Huntington là một rối loạn di truyền nguy hiểm ảnh hưởng đến chức năng vận động, nhận thức và tâm thần.

Bệnh Huntington là một rối loạn thần kinh tiến triển âm thầm nhưng để lại những tác động sâu rộng lên vận động, tư duy và cảm xúc của người mắc.
Từ các cử động không kiểm soát đến suy giảm nhận thức và thay đổi hành vi, căn bệnh này không chỉ ảnh hưởng đến cá nhân người bệnh mà còn đặt ra nhiều thách thức cho gia đình và người chăm sóc.
Bệnh Huntington là gì?
Bệnh Huntington, còn được gọi là bệnh múa giật, là một rối loạn thoái hóa thần kinh di truyền hiếm gặp. Bệnh gây tổn thương tiến triển ở não bộ, dẫn đến suy giảm dần chức năng vận động, nhận thức và tâm thần. Đây là bệnh lý mạn tính, diễn tiến theo thời gian và hiện chưa có phương pháp điều trị khỏi hoàn toàn.
Tên gọi “bệnh múa giật” xuất phát từ một biểu hiện vận động đặc trưng của bệnh, được gọi trong y học là chorea. Đây là các cử động nhanh, không tự chủ, xảy ra ở tay, chân, mặt hoặc thân mình, khiến người bệnh trông như đang thực hiện các động tác múa nhẹ.
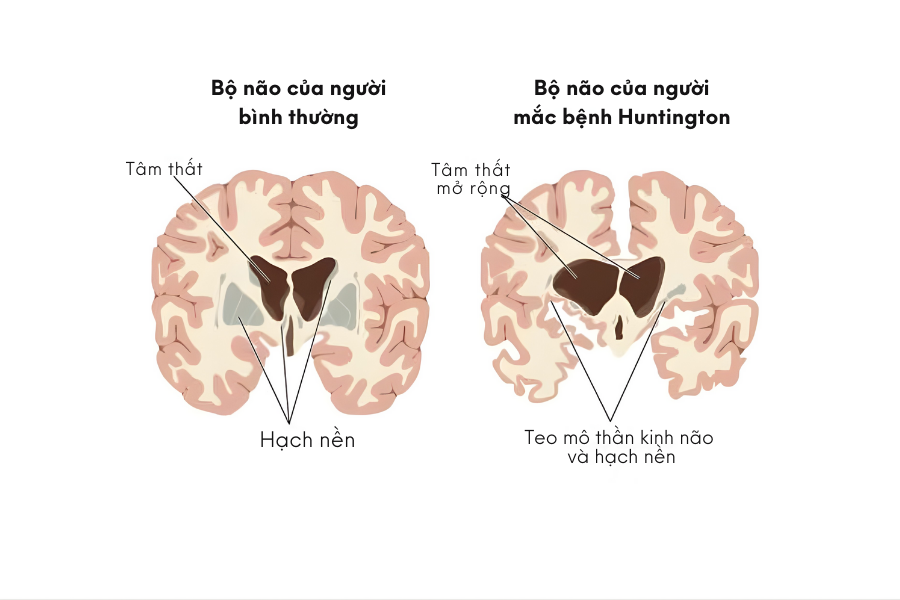
Ngoài rối loạn vận động, bệnh Huntington còn ảnh hưởng đến trí nhớ, khả năng suy nghĩ, cảm xúc và hành vi, với mức độ khác nhau ở từng người bệnh. Các triệu chứng này thường xuất hiện từ từ và nặng dần theo thời gian.
Bệnh Huntington thường khởi phát ở độ tuổi trung niên, phổ biến nhất trong khoảng từ 30 đến 50 tuổi, dù vẫn có những trường hợp xuất hiện sớm hơn.
Triệu chứng của bệnh Huntington và những ảnh hưởng của nó đến đời sống
Bệnh Huntington là một rối loạn thần kinh tiến triển, trong đó các triệu chứng xuất hiện từ từ và nặng dần theo thời gian. Những biểu hiện này không chỉ ảnh hưởng đến sức khỏe thể chất mà còn tác động sâu sắc đến đời sống tinh thần, khả năng lao động và các mối quan hệ xã hội của người bệnh.
Mức độ và tốc độ tiến triển có thể khác nhau giữa từng cá nhân, nhưng nhìn chung bệnh gây suy giảm toàn diện theo thời gian.
Bên cạnh đó, do tính chất kéo dài, căn bệnh này còn ảnh hưởng lớn đến gia đình và người chăm sóc, cả về tâm lý lẫn kế hoạch chăm sóc lâu dài.
Rối loạn vận động
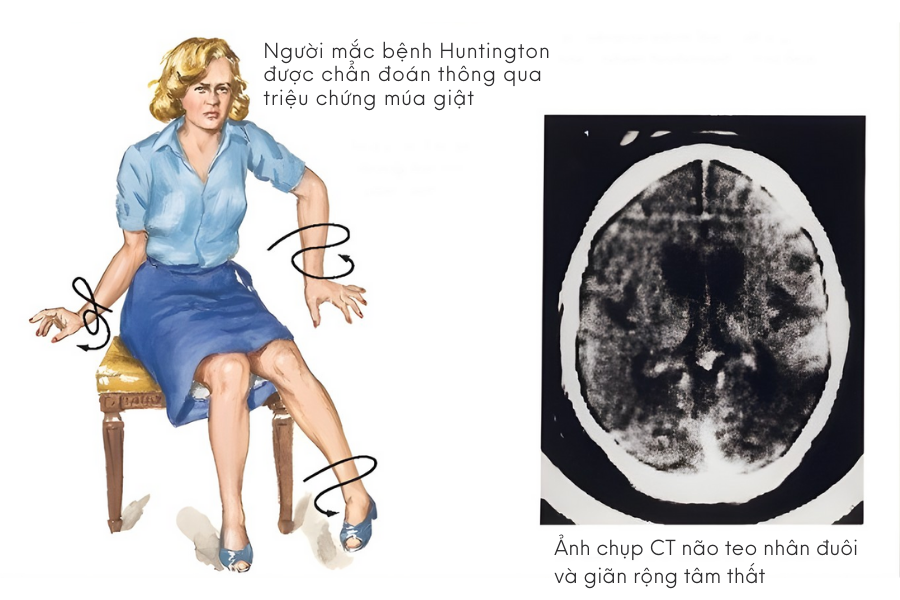
Một trong những biểu hiện đặc trưng của bệnh Huntington là các cử động không tự chủ. Những cử động này thường xảy ra ở ngón tay, bàn chân, mặt hoặc thân mình, khiến người bệnh trông như đang cử động liên tục. Múa giật có xu hướng rõ hơn khi người bệnh lo lắng hoặc mất tập trung và có thể trở nên nặng dần khi bệnh tiến triển.
Các triệu chứng của bệnh Huntington thường khởi phát ở tuổi trung niên, nhưng cũng có thể xuất hiện ở trẻ em hoặc thanh thiếu niên, dù trường hợp này hiếm gặp. Bệnh có tính chất tiến triển, các biểu hiện ngày càng nặng hơn theo thời gian.
Ở giai đoạn sớm, người bệnh có thể chỉ gặp những thay đổi nhẹ như vụng về, mất thăng bằng, gặp khó khăn khi thực hiện những cử động nhỏ, chính xác và có chủ đích, kèm theo các rối loạn về tư duy, cảm xúc hoặc hành vi.
Không phải tất cả người mắc Huntington đều biểu hiện múa giật rõ ràng. Ở một số trường hợp, người bệnh có thể bị cứng cơ, giảm vận động hoặc gần như không vận động được, tình trạng này được gọi là giảm vận động (akinesia).
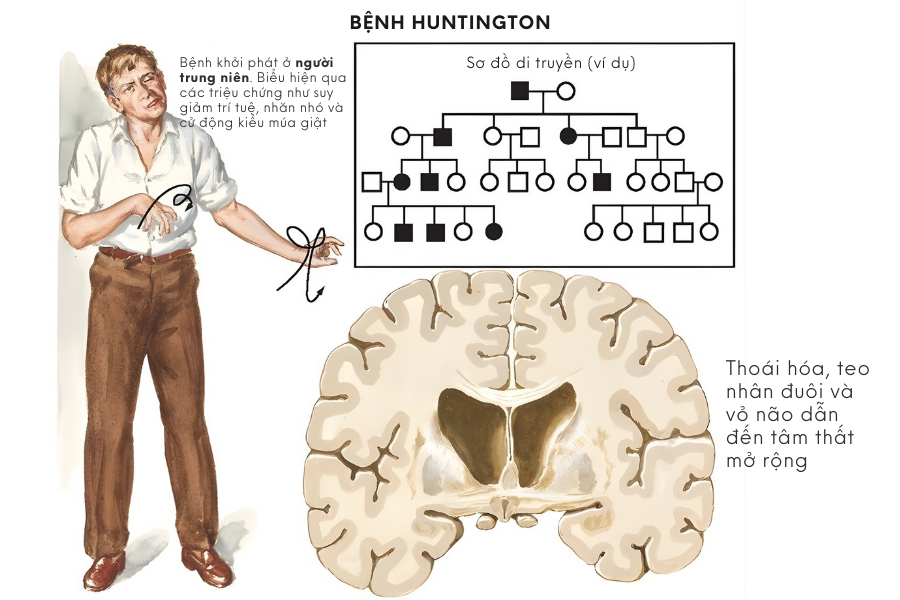
Một số người ban đầu có múa giật nhưng về sau lại chuyển sang cứng cơ khi bệnh tiến triển. Ngoài ra, người bệnh có thể xuất hiện tư thế bất thường kéo dài, được gọi là loạn trương lực cơ (dystonia). Các rối loạn vận động này có thể xen kẽ hoặc đồng thời tồn tại.
Những biểu hiện vận động khác có thể bao gồm run và các bất thường trong chuyển động mắt, trong đó rối loạn vận nhãn có thể xuất hiện sớm.
Khi bệnh tiến triển, người bệnh thường gặp khó khăn trong việc nói, nuốt, ăn uống và đi lại. Những vấn đề này làm tăng nguy cơ sụt cân, nghẹn, nhiễm trùng đường hô hấp và suy kiệt thể chất.
Ngoài ra, người bệnh có thể gặp tình trạng mất ngủ, mệt mỏi kéo dài, giảm năng lượng và trong một số trường hợp có thể xuất hiện co giật. Ở giai đoạn muộn, nhiều người cần phải nằm liệt giường hoặc sử dụng xe lăn.
Suy giảm khả năng tư duy và xử lý thông tin
Người bệnh có thể gặp khó khăn trong việc tập trung, phán đoán, giải quyết vấn đề và đưa ra quyết định. Những rối loạn này ảnh hưởng trực tiếp đến khả năng học hỏi, ghi nhớ, sắp xếp công việc, diễn đạt suy nghĩ và phản hồi trong giao tiếp.
Theo thời gian, suy giảm nhận thức trở nên nghiêm trọng hơn, khiến người bệnh không còn khả năng làm việc, lái xe hoặc tự chăm sóc bản thân. Khi suy giảm nhận thức ảnh hưởng nghiêm trọng đến khả năng sinh hoạt hằng ngày, tình trạng này được mô tả là sa sút trí tuệ.
Tuy vậy, nhiều người mắc bệnh Huntington vẫn có thể nhận biết môi trường xung quanh và bộc lộ cảm xúc, ngay cả ở giai đoạn muộn.

Thay đổi về hành vi và cảm xúc
Người bệnh có thể trở nên dễ cáu gắt, thay đổi tâm trạng, giảm hứng thú với các hoạt động thường ngày, hoặc cảm thấy thờ ơ, buồn bã và tức giận.
Ở một số trường hợp, các triệu chứng này có thể giảm dần theo thời gian, nhưng cũng có người tiếp tục gặp các rối loạn nghiêm trọng hơn như bùng phát cơn giận dữ, trầm cảm nặng, ý nghĩ tự tử hoặc rối loạn tâm thần.
Những thay đổi này thường khiến người bệnh thu mình khỏi các hoạt động xã hội và làm tăng gánh nặng cho gia đình, người chăm sóc.
Các triệu chứng tâm thần trong bệnh Huntington đôi khi xuất hiện trước các biểu hiện vận động, khiến việc chẩn đoán sớm trở nên khó khăn hơn.



Nguyên nhân và cơ chế di truyền của bệnh Huntington
Bệnh Huntington là một rối loạn di truyền, trong đó đột biến ở một gen nhất định được truyền từ cha hoặc mẹ sang con.
Nguyên nhân của bệnh Huntington
Nguyên nhân trực tiếp của bệnh là do đột biến gen HTT, nằm trên nhiễm sắc thể số 4. Gen HTT có vai trò tạo ra protein huntingtin, một loại protein cần thiết cho sự phát triển và hoạt động bình thường của tế bào thần kinh.
Khi gen này bị đột biến, protein huntingtin được tạo ra sẽ mang đặc tính bất thường và gây tổn thương cho các tế bào thần kinh trong não.
Đột biến gen HTT liên quan đến sự gia tăng bất thường của một đoạn lặp lại trong cấu trúc gen, được gọi là đoạn lặp CAG. Ở người bình thường, số lần lặp CAG nằm trong giới hạn an toàn.
Khi số lần lặp này vượt quá ngưỡng nhất định, protein huntingtin trở nên độc hại đối với tế bào thần kinh. Sự tích tụ của protein bất thường này dẫn đến rối loạn chức năng và thoái hóa dần các tế bào thần kinh, đặc biệt tại những vùng não liên quan đến kiểm soát vận động, nhận thức và hành vi.
Chính sự mất dần tế bào thần kinh ở các vùng não này là nguyên nhân gây ra các triệu chứng đặc trưng của bệnh Huntington.
Cơ chế di truyền
Bệnh Huntington được truyền theo kiểu trội trên nhiễm sắc thể thường. Điều này có nghĩa là chỉ cần một bản sao gen HTT bị đột biến từ cha hoặc mẹ, người con đã có nguy cơ mắc bệnh.
Nếu một người mang gen Huntington, mỗi người con của họ có khoảng 50% khả năng thừa hưởng gen đột biến này. Cơ chế di truyền trội khiến bệnh Huntington có thể xuất hiện liên tiếp qua nhiều thế hệ trong cùng một gia đình.
Trong một số trường hợp hiếm gặp, bệnh Huntington có thể được chẩn đoán ở người không ghi nhận tiền sử gia đình rõ ràng. Các tài liệu y khoa cho thấy điều này có thể liên quan đến sự gia tăng số lần lặp CAG trong quá trình di truyền qua các thế hệ, đặc biệt khi gen được truyền từ người cha sang con. Tuy nhiên, cơ chế này vẫn đang tiếp tục được nghiên cứu.
Phương pháp chẩn đoán bệnh Huntington
Xét nghiệm di truyền có thể xác định chính xác số lần lặp CAG trong gen HTT và được xem là phương pháp tiêu chuẩn để xác nhận nguy cơ và chẩn đoán bệnh Huntington.
Do tính chất di truyền rõ ràng và tác động lâu dài, việc tư vấn di truyền đóng vai trò quan trọng đối với những người có tiền sử gia đình mắc bệnh Huntington. Tư vấn này giúp cá nhân và gia đình hiểu rõ nguy cơ, hệ quả sức khỏe và những vấn đề tâm lý có thể phát sinh khi đối mặt với một bệnh lý di truyền tiến triển.
Bên cạnh đó, bác sĩ thần kinh sẽ đánh giá chức năng vận động, nhận thức và tâm thần của người bệnh thông qua các bài kiểm tra chuyên biệt. Chụp cộng hưởng từ hoặc chụp cắt lớp não có thể được sử dụng để quan sát những thay đổi cấu trúc não, dù không phải là tiêu chuẩn chẩn đoán duy nhất.
Phương pháp điều trị bệnh và quản lý bệnh Huntington
Hiện tại, y học chưa có phương pháp điều trị giúp loại bỏ hoàn toàn nguyên nhân gây bệnh Huntington. Các phương pháp điều trị chủ yếu tập trung vào kiểm soát triệu chứng và cải thiện chất lượng sống cho người bệnh.
Một số loại thuốc có thể được sử dụng để giảm các cử động không kiểm soát hoặc hỗ trợ điều trị rối loạn tâm thần như trầm cảm và lo âu.
Song song đó, vật lý trị liệu, hoạt động trị liệu và ngôn ngữ trị liệu đóng vai trò quan trọng trong việc duy trì khả năng vận động, giao tiếp và sinh hoạt hằng ngày.
Những tiến bộ nghiên cứu mang lại hy vọng mới
Năm 2025 được xem là một cột mốc đáng chú ý trong nghiên cứu bệnh Huntington, khi các dữ liệu ban đầu từ thử nghiệm lâm sàng cho thấy thuốc AMT-130 có khả năng làm chậm tiến triển của bệnh.
AMT-130 là một phương pháp điều trị đang được nghiên cứu, hướng đến việc can thiệp trực tiếp vào cơ chế sinh bệnh thay vì chỉ tập trung kiểm soát các triệu chứng như trước đây.
Liệu pháp này sử dụng một loại vi rút vô hại để đóng vai trò là phương tiện đưa một đoạn DNA đã được chỉnh sửa riêng vào các tế bào thần kinh trong não.
Khi đoạn DNA tiếp cận và đi vào tế bào thần kinh, nó tham gia điều chỉnh hoạt động của gen HTT, từ đó làm giảm việc sản xuất protein huntingtin có hại, yếu tố được xem là trung tâm trong cơ chế bệnh sinh của Huntington.

Để hạn chế nguy cơ phản ứng nghiêm trọng, vi rút mang DNA được truyền vào não với tốc độ rất chậm thông qua một ống thông siêu nhỏ. Thuốc được đưa trực tiếp vào hai vùng não xác định trong một ca phẫu thuật thần kinh kéo dài, thường từ 12 đến 20 giờ.
Kết quả thử nghiệm ban đầu, thu thập từ 29 bệnh nhân tham gia điều trị tại Anh và Mỹ, đã được công bố bởi công ty công nghệ sinh học uniQure của Hà Lan. Theo dữ liệu này, tốc độ tiến triển bệnh ở nhóm bệnh nhân được điều trị bằng liều cao chậm hơn rõ rệt so với các nhóm còn lại.
Các chỉ số sinh học cũng cho thấy mức độ tổn thương và chết tế bào thần kinh ở những người tham gia điều trị thấp hơn.
*Nội dung bài viết được biên soạn nhằm mục đích cung cấp thông tin sức khỏe hữu ích, không thay thế cho chẩn đoán hay điều trị y khoa. Nếu bạn có vấn đề về sức khỏe, hãy tham khảo ý kiến bác sĩ chuyên khoa.
Tóm lại
1. Bệnh Huntington là gì?
Bệnh Huntington là một rối loạn thần kinh di truyền hiếm gặp, gây thoái hóa tiến triển các tế bào thần kinh trong não. Bệnh ảnh hưởng đến vận động, tư duy và cảm xúc, thường khởi phát ở tuổi trung niên và nặng dần theo thời gian.
2. Những triệu chứng của bệnh Huntington?
Bệnh Huntington gây ra các cử động không tự chủ như múa giật, rối loạn thăng bằng và khó nói, khó nuốt. Ngoài ra, người bệnh còn suy giảm trí nhớ, khó tập trung, thay đổi cảm xúc và hành vi, trong đó trầm cảm và dễ cáu gắt khá phổ biến.
3. Nguyên nhân và cơ chế di truyền của bệnh Huntington là gì?
Bệnh Huntington do đột biến gen HTT trên nhiễm sắc thể số 4, khiến cơ thể tạo ra protein huntingtin bất thường gây tổn thương tế bào thần kinh. Bệnh di truyền theo kiểu trội, nghĩa là chỉ cần thừa hưởng gen đột biến từ cha hoặc mẹ là đã có 50% nguy cơ mắc bệnh.
4. Phương pháp điều trị hiện nay của bệnh Huntington?
Hiện chưa có phương pháp điều trị khỏi bệnh Huntington. Việc điều trị chủ yếu tập trung vào kiểm soát triệu chứng bằng thuốc, kết hợp vật lý trị liệu, ngôn ngữ trị liệu và hỗ trợ tâm lý nhằm cải thiện chất lượng sống.